Pairk - tutorial
introduction
Pairk is a Python package for quantifying the conservation of motifs within intrinsically disordered regions (IDRs). It will run on any input protein sequences that you provide but it is designed for disordered regions, where multiple sequence alignments are particularly difficult to interpret and unreliable
the pairk method can be split into 2 steps, where each step has configurable options:
Step 1: pairwise k-mer alignment - aligns all of the k-mers in the
query sequence to all of the k-mers in the homolog sequences. For each
query k-mer, the best matching k-mer from each homolog is selected and
recorded. The results are returned in a PairkAln object which
provides methods for accessing the results and writing/reading the
results to/from files. - The way in which the best matching query k-mer
- homolog k-mer match is selected is adjustable. The currently
implemented methods use either scoring matrices to score the match or
use residue embedding distances from the ESM2 protein language model to
score the match. Both methods are described in detail below
Step 2: k-mer conservation - calculates conservation scores for each
query k-mer using the results of Step 1. The conservation score results
are stored in a PairkConservation object which also provides methods
for plotting the results as well as writing/reading the results to/from
files. - The function used to score the conservation is adjustable.
make sure that pairk is installed before running this notebook (see the README.md file for installation instructions)
import
Most of the functionality of pairk is accessed through the main pairk module, which can be imported directly.
import pairk
load example data
pairk comes with some example data that we can use to test the methods.
The example data is stored in the pairk.example1 object.
ex1 = pairk.example1
This example is imported as a python object and holds data that is compatible with the main pairk methods
for example, we can access the IDR sequences in a dictionary with
ex1.idr_dict or the query id with ex1.query_id
for id, seq in ex1.idr_dict.items():
print(id, seq)
9606_0:00294e TNLGTVNAAAPAQPSTGPKTGTTQPNGQIPQATHSVSAVLQEAQRHAETSKDKKPALGNHHDPAVPRAPHAPKSSLPPPPPVRRSSDTSGSPATPLKAKGTGGGGLPAPPDDFLPPPPPPPPLDDPELPPPPPDFMEPPPDFVPPPPPSYAGIAGSELPPPPPPPPAPAPAPVPDSARPPPAVAKRPPVPPKRQENPGHPGGAGGGEQDFMSDLMKALQKKRGNVS
9793_0:005123 TNVGTGNAATPAPPSTGLKTGTAQANGQIPQAAHSVSTVLNEADRQVDTPKDKKPALSNHDPGTPRAQHLPKSSLPPPPPVRRSSDTSSSPVMPAKGAAGGLPPLLDDSLPPPPPPPPLEDDELPPPPPDFDDAPPNFVPPPPPWDAGASLPPPPPPPPPALALAPEATKPSPVVAKRPPVPPKRQENPAPASGGGGGEQDFMSDLMKALQKKRGNVA
1706337_0:000fc7 TNLGTVNAAPPAPSSTGVKTGTTQANGQIPQAAHSMSTVLGEAQRQVETTKDKKSGLGSHDPGAPRAQTLPKSSLPPPPPVRRSSEVGCGSPGTSPKVKGAAAGFPAPPHDLLPPPPPPPPLEDDELPPPPPDFSDAPPDFVPPPPPPSFAGDAGSSLPPPPPPPALAPEAAKPTPVVVKRPPAPPKRQANPGPPGGGGGEQDFMSDLMKALQKKRSNMP
51337_0:001b5a TNLGTVNTATPAQPSTGFKTGSSQPNGQIPQTIPSVSAGLQEAQRHETIKDKKPSLSSTEPGAPRDPPGARSSLPPPPPPVRRSSDTCARAASPFPAPPDDLPPPPPPPPLEDPAMLPPPPALPEPPPDCVPPPPPPPGPGPQPARPSPGAGRRPPVPPKRQENPGLPSAGAGGEQDFMSDLMKALQKRGHMP
9568_0:004ae1 TNLGTVNAAAPAQPSTGPINGTAQPNGQMPQAAHSVSAVLQEAQRHAETSKVKPARPINGTAQPNGQMPQAAHSVSAVLQEAQRHAETSKRPSPAVAKRPPMPPKRHENPGTPSGAGGGEQDFMSDLMKALQKKRGNVS
43346_0:004190 TNLGTVNAVPPAPPSTGVKTGTTQANGQLPQATQSMNTALGEDWRQLETTKDKKPGPGLGSHDPGAPRAQPLPKSSLPPPPPPVRRSSDVGGAPPPSFAEDLPPPPPPPPALAPESVRTPPVVVKRPPPPPKRQENPGPPGGGGGEQDFMSDLMKALQKKRGNVS
885580_0:00488c TNLGTVNTAAAAQPPAGLRTGTSQPNGQLPRDAPCLSDALRESQRQADTSKVGPEGQGGHELPDRPKAAFQASTALAASPYSPKH
10181_0:00305d TNLGTVSTATAAQPPTGLRTSTTQHNGQLPQAAPHLSAVLQEAQRQAEASKVGPEGNRPKLQQISAPSPPSKPEAVFWHLLLFPASENPPYCNFT
1415580_0:000900 PNLSKVDPTVTARSSSSGAVQANGQIHQNVIPVISTNPEAFKRAEDKKPNVGRKPDQAQLQPVPSSNHQQSPKVALHASKIPPPAPARISSQAYSSALTLPSNVKNVNANVLLPPPPSPSPPPPDAFPLPPPCNNDLPPPPDDFYDPPPDFLPPPPPCFATGDRAQLPPGPPLPPPPPSSNQPKPFMKKPVPLPPKRQDITSLHSEQPSLAGPTPVGGGGGQPDFMSDLMKALQKKRGSTS
61221_0:00105a AKSGNTESIVAVGTSAKGSTHANGQVPQSITLSKTDSSETGKSAEMPKVKKPDNTADSIQAPTPNTPQLKHQRKAGGSHSAPPMPPQRVSSAVTAPLQLPTNAEGKGKVCPSDAAEFPPPPESMLPPPELEDLPLPPPPPPEYFESPPDFIPPPPPSCAVAVSAGAPPLPPPPPSASLPRMPLSIKKKPPPPPRRQEESAGQAGLPKPSAPPPKTETAGQGDFMSDLMKALEKKRGATS
7897_0:0033c5 ANFAKVQSQNTTASSTGSVQANGHAGQVTPVSVSFSEAWKRGDVGKEKQSNDGQDNLPPPPPPPPPPMQGFMNEAFPPPPSLPPIASGSLPPPLRASASAPAPPPISNNFPPPLDELSPPPDDFDFPEPPPDFLPPPPTVSASGVPPPPPPPPPPPAPTAASQPTPLPKKSVPPRRQENTTLSQPRGGGGGGQPDFMSDLTKALQKKRGNAS
8407_0:002bff SSLSSSTTPTGAPQGNGQISQNVANVSSSFSDAWKRGETAKDKQQPTEVRKPEQKISLPPSTKQPPPAPVRRPSNAHVVGTPPLPIKAKPVTSNMPPPPPPAEASQWGDDFLPPPPPPELLDTPPNFLPPPPPSFNSESDYPAPPQFTNVGSAGGPPPPPPPPPPPPAALSPKSAPPQLPVKKLPPKPPMRRDSTGQRPNQQNSLMTNGGGAGGQPDFMSDLMSALQKKRSTTT
173247_0:004550 MNRSTPSSSNPSTPSPTIKAKTPNQANGHAPKPQPTAPDSMDFGNFPPPPSADILPPPPPDPAFPPPPPSLPAKSSSRPVAPQHKLPANFPPPPMAMDNLPPPPLPPPIDDSPEAPPDFLPPPPPAAGFGSLPPPPLSMNSLPPPPHFGGMDQSLPPPPPDPEFLPPPPPEPVFTGAGAPPPPPPPPPPPPAQAAAVPRAPVRPSGSVRKVPPAPPKRTTPSLQVGGGGGGGDFMSELMVAMQKKRGDH
30732_0:0046dd TNRSTPSSSNPSTPSPTVKAKAQSQANGHAPKPQPGPVSQDFGHLPPPPPPCPNDDLPPPPPDPVFPPPPPPLAAKRSPKTAGRSQHPQGNFPPPPPEMDHLPPPPPMEESPPDFLPPPPPMNSLPHPPPPPASFGGVDHSLPPPPPDPEFLPPPPPDPQVTGGGGPPPPPPPPPPPPPASAPAPRGALRPTGSAKKMPPAPPKRTTPVMGGGGGGGGGGGGDFMSELMKAMQKKRSDQ
241271_0:0048e4 TNRTPVSSNQSTPSPTIKAKSPNQANGHAAKPQPGPESQDFGNIPPPPPPPPPPMTGFLPPPPPDPVLPPPPPLLAAKSPKPSPPQRNLPTNFPPPAMDNLPPPPPPPMDDSFEDPPDFLPPPPPAAGFGSLPPPPPPVNSFPPPPPSAGFGGMGQSLPPPPPDPGFLPPPPPQPMFTGGGTIPPPPPPPPPPTAAPRAPVRPTGSVKKAPPAPPKRTTPSLHGGGGGGGGGGGDFMSELMMAMNKKRGTT
8103_0:0045e4 TNRSTPSSSNPSTPSPPIKAKSPGQANGHALKPEPGPVAQDFGHVPPPPPADILPPPADILPPPPPQTFLPPPPPPLAAKSSSKPSLPQRHLPTNFPPPPPAMINLPRPPQPPPTDDASEAPPDFLPPPPPAAGFSPLFPPPPPLNALPLPPPPVSFRVEDRSLPPPPPDPGFLPPPPPMFTGAGAPPPPPPPPPPPRVAVRPAGSVKKRPPAVPKRTTPSLRGGGGGDFMSELALAMNKKRSAH
56723_0:00152f TNRSTPSSSSSSTPSPTIKAKSPSQANGHAPKPQPGPVSQDFGNVPPPPPPMANILPPPRPDAFLPPAPPPLARKNSAKPPPPQRHLPTNFPPPPPAMDNLPPPPPPPPMDDALEAPPDFLPPPPPAAGFGSLPPPPPPSNSFPPPPPPGSFGSMGQSLPPPPPDPGFLPPPPPQPVFTGAGAPPPPPPPPPPPTAAAAPRAPVRPSGSVKKIPPATPKRTTPSLQGGGGGGGGGGGGGGDFMSELMLAMNKKRST
210632_0:004c0c ANRSTSSSSNSSTPSPTIKAKSSSQANGIFPKPGPAPQDFGDLPPPPPLAANILPPPPPEPGLPPPPPPPPPQAAKGSAKPAPPKRQMPANFPPPPTAMDNLPPPPPPPPIDNSEAPPDFLPPPPPASGFGSFPPPPPLNSLPPPPRPGGFGGMDQSLPPPPPDPEFLPPPPPPPQAVFTGGGAPPPPPPPPPPPAAAAPSTAIPRVGLRPAGSLKKLPPAPPKRTTPSMQGSGGGGGGDFMSELMLAMQKKRGDHP
31033_0:00264e TNCSKPSSDGPPTPPTTIKASPANGHVPKPPPGAAPQDVFPPPPPPMDILPPPPPDPAFPPPPPPLMAKRSPKPSAGHRQAPGDHLPPPPLAPPHDDASEDPPDFLPPPPPSFDSLPPPPPGMSAFPPPPPLLGFSETSQPLPPPPPDPELLLPPPPASMISTGAGAPPPPPPPPPAAAASPRPAPTASGSVRKRPPAPPKRTTPALHGSGGGAGEGAGGGDFMSELMKAMNKKRADHS
63155_0:004c86 TNRSTPSSSNQSTPSPTVKAKSPNQANGHPPKPQPGPISQAPFPPPPLAEVLPPPPPDPVLPPPPPMPAKSSAKPSPPKRQQQSNFPPPPPELDNLPPPPPPPPTDDTAEAPPDFLPPPPPAVGFGSLPPPPPSFGGVGQSLPPPPPDPQSLPPPPPDPVFIGAGAPPPPPPPPPPPAPGAPVTTLRPAVRPSGSLKKVPPAPPQRNTPSVSGGGGGGGGDFMSELMLAMQKKRGAQ
7994_0:004d71 TSMNRQPSPSTSNTSTPSPTPKAKTANGHASQPRSETVPKAPSNQSAFPPPPPPADFLPPPPPDPTLPPPPPPPPALPVKKESNPPRSAPQRSQPAFPPPPPAMDFSLPPPPPPSDDLEMPPDFLPPPPPAPGGFMGGDLLPPPPPEPFHAPLPPPPAAFHPPPAVHPPPQATGGDLPPPPPPPPPPPPAPAAFHQTPSVRKVGPPPPKRTTPSLAAPSGGDFMSELMLAMNKKRGGQ
109280_0:00369f ASCSVTSGQKQSQSQVANGHANNTSPSPLPPPPPPLGEDLPPPPPPPPQLGKTLPPAPPPLGATLPTPPPPPGGTPPPPPPPRRNPPPYPRHLPHISELYPLRRLLLLYQLPTVHSLVLLFQPNLPPNPSPNHDVQRPISRCLPGPQITFPPPPPPPVDDSPPDFLPPPPPAANFGSHPPPPPPVKTLPPPPPHMKTLPPARLSFKSTNLPPPPPDPGFLPPPLTGVPPPPPPPPPPPPTTAAAGPRRAPVRPSGSLKKMPPPPPKRSTPSLHGRRDGDRGDGDGGGGGDFMSELMRAMQKKRDPH
150288_0:004e5a HPPDRCSTSSDNLNSQIGQSAPTPDECIEQDEPPPDFIPPPPPGYMAIL
print(ex1.query_id)
9606_0:00294e
Step 1: pairwise k-mer alignment
there are 2 main ways to run the k-mer alignment step:
Scoring matrix alignment
These methods use a scoring matrix to score the query k-mer to homolog k-mer matches and select the best scoring match from each homolog.
Embedding distance alignment
This method uses the Euclidean distance between the query k-mer residue embeddings from a protein large language model (such as ESM2) and homolog k-mer residue embeddings and selects the lowest distance match from each homolog.
We will only go through one of the scoring matrix alignment methods in this tutorial. They are all described in detail in PairK alignment - in depth
k-mer alignment - pairk.pairk_alignment()
an exhaustive comparison of all k-mers in the query sequence with all k-mers in the homologs. Uses a scoring matrix to score the query k-mer to homolog k-mer matches and select the best scoring match from each homolog.
The pairk.pairk_alignment() function takes the following arguments:
idr_dict: a dictionary of IDR sequences, where the keys are the sequence ids and the values are the sequences. Includes the query sequence (the sequence to split into k-mers and align with the homologs).query_id: a query sequence id (the sequence to split into k-mers and align with the homologs). This id should be present inidr_dict.k: the length of the k-mers
These inputs are provided by the user. There are helper functions
in the pairk library that might help with this (see functions in
pairk.utilities).
aln_results = pairk.pairk_alignment(
idr_dict=ex1.idr_dict,
query_id=ex1.query_id,
k=5,
)
To specify the scoring matrix used, you can pass the name of the matrix
to the matrix_name argument.
To see the available matrices, use the
pairk.print_available_matrices() function.
pairk.print_available_matrices()
biopython-builtin matrices (aligner compatible):
BENNER22
BENNER6
BENNER74
BLASTN
BLASTP
BLOSUM45
BLOSUM50
BLOSUM62
BLOSUM80
BLOSUM90
DAYHOFF
FENG
GENETIC
GONNET1992
HOXD70
JOHNSON
JONES
LEVIN
MCLACHLAN
MDM78
MEGABLAST
NUC.4.4
PAM250
PAM30
PAM70
RAO
RISLER
SCHNEIDER
STR
TRANS
other matrices:
grantham_similarity_normx100_aligner_compatible
BLOSUM62
EDSSMat50
grantham
grantham_similarity_norm
k-mer alignment results - pairk.PairkAln
The results of all of the pairwise k-mer alignment methods are returned
as a pairk.PairkAln object.
The actual “alignments” are stored as matrices in the pairk.PairkAln
object. The main matrices are:
orthokmer_matrix- the best matching k-mers from each homolog for each query k-merposition_matrix- the positions of the best matching k-mers in the homologsscore_matrix- the scores of the best matching k-mers
Each matrix is a pandas DataFrame where the index is the start position of the k-mer in the query sequence. The columns are the query k-mers + the homolog sequence ids.
The pairk.PairkAln object has some useful methods for accessing the data.
For example, you can get the best matching k-mers for a query k-mer by
its position in the query sequence using the pairk.get_pseudo_alignment()
method (or by directly accessing the dataframes). You can also plot the
matrices as heatmaps, save the results to a json file, and load the
results from that file
example: accessing the DataFrames from the pairk.PairkAln object directly
aln_results.score_matrix
| query_kmer | 9793_0:005123 | 1706337_0:000fc7 | 51337_0:001b5a | 9568_0:004ae1 | 43346_0:004190 | 885580_0:00488c | 10181_0:00305d | 1415580_0:000900 | 61221_0:00105a | ... | 30732_0:0046dd | 241271_0:0048e4 | 8103_0:0045e4 | 56723_0:00152f | 210632_0:004c0c | 31033_0:00264e | 63155_0:004c86 | 7994_0:004d71 | 109280_0:00369f | 150288_0:004e5a | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | TNLGT | 22.0 | 28.0 | 28.0 | 28.0 | 28.0 | 28.0 | 28.0 | 10.0 | 9.0 | ... | 13.0 | 12.0 | 13.0 | 13.0 | 9.0 | 8.0 | 13.0 | 8.0 | 14.0 | 9.0 |
| 1 | NLGTV | 16.0 | 29.0 | 29.0 | 29.0 | 29.0 | 29.0 | 29.0 | 16.0 | 8.0 | ... | 11.0 | 13.0 | 11.0 | 13.0 | 7.0 | 7.0 | 11.0 | 6.0 | 14.0 | 10.0 |
| 2 | LGTVN | 16.0 | 29.0 | 29.0 | 29.0 | 29.0 | 29.0 | 23.0 | 10.0 | 7.0 | ... | 11.0 | 8.0 | 9.0 | 10.0 | 7.0 | 8.0 | 10.0 | 8.0 | 16.0 | 4.0 |
| 3 | GTVNA | 19.0 | 26.0 | 23.0 | 26.0 | 26.0 | 23.0 | 17.0 | 15.0 | 10.0 | ... | 11.0 | 9.0 | 10.0 | 8.0 | 8.0 | 9.0 | 11.0 | 9.0 | 9.0 | 6.0 |
| 4 | TVNAA | 18.0 | 25.0 | 22.0 | 25.0 | 22.0 | 22.0 | 16.0 | 14.0 | 11.0 | ... | 11.0 | 9.0 | 8.0 | 12.0 | 8.0 | 11.0 | 11.0 | 10.0 | 11.0 | 5.0 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 217 | LQKKR | 31.0 | 31.0 | 19.0 | 31.0 | 31.0 | 3.0 | 9.0 | 31.0 | 24.0 | ... | 26.0 | 19.0 | 19.0 | 19.0 | 26.0 | 19.0 | 26.0 | 19.0 | 26.0 | 0.0 |
| 218 | QKKRG | 29.0 | 23.0 | 16.0 | 29.0 | 29.0 | 5.0 | 3.0 | 29.0 | 22.0 | ... | 23.0 | 22.0 | 16.0 | 16.0 | 29.0 | 17.0 | 29.0 | 22.0 | 23.0 | -1.0 |
| 219 | KKRGN | 29.0 | 23.0 | 18.0 | 29.0 | 29.0 | 2.0 | 2.0 | 23.0 | 21.0 | ... | 17.0 | 23.0 | 15.0 | 17.0 | 23.0 | 18.0 | 21.0 | 22.0 | 14.0 | -1.0 |
| 220 | KRGNV | 29.0 | 19.0 | 20.0 | 29.0 | 29.0 | 8.0 | 8.0 | 17.0 | 15.0 | ... | 9.0 | 17.0 | 8.0 | 11.0 | 14.0 | 9.0 | 13.0 | 14.0 | 11.0 | 2.0 |
| 221 | RGNVS | 23.0 | 12.0 | 13.0 | 27.0 | 27.0 | 9.0 | 13.0 | 15.0 | 13.0 | ... | 10.0 | 11.0 | 8.0 | 13.0 | 7.0 | 9.0 | 7.0 | 9.0 | 8.0 | 9.0 |
222 rows × 23 columns
example: access the best matching k-mers for the query k-mer at position 4:
print(aln_results.orthokmer_matrix.loc[4])
query_kmer TVNAA
9793_0:005123 TGNAA
1706337_0:000fc7 TVNAA
51337_0:001b5a TVNTA
9568_0:004ae1 TVNAA
43346_0:004190 TVNAV
885580_0:00488c TVNTA
10181_0:00305d TVSTA
1415580_0:000900 NVNAN
61221_0:00105a AVSAG
7897_0:0033c5 TVSAS
8407_0:002bff SQNVA
173247_0:004550 TPNQA
30732_0:0046dd TVKAK
241271_0:0048e4 PVNSF
8103_0:0045e4 PLNAL
56723_0:00152f TAAAA
210632_0:004c0c TIKAK
31033_0:00264e TIKAS
63155_0:004c86 TVKAK
7994_0:004d71 TSNTS
109280_0:00369f TTAAA
150288_0:004e5a NLNSQ
Name: 4, dtype: object
example: access the best matching k-mers for the query k-mer at position
4 using the get_pseudo_alignment method. (the returned list includes
the query k-mer sequence)
aln_results.get_pseudo_alignment(4)
['TVNAA',
'TGNAA',
'TVNAA',
'TVNTA',
'TVNAA',
'TVNAV',
'TVNTA',
'TVSTA',
'NVNAN',
'AVSAG',
'TVSAS',
'SQNVA',
'TPNQA',
'TVKAK',
'PVNSF',
'PLNAL',
'TAAAA',
'TIKAK',
'TIKAS',
'TVKAK',
'TSNTS',
'TTAAA',
'NLNSQ']
you can search for a specific kmer to get its positions. You can then use the positions to query the matrices.
aln_results.find_query_kmer_positions('LPPPP')
[75, 113, 127, 157]
aln_results.get_pseudo_alignment(75)
['LPPPP',
'LPPPP',
'LPPPP',
'LPPPP',
'PPMPP',
'LPPPP',
'LPDRP',
'APSPP',
'LPPPP',
'LPPPP',
'LPPPP',
'LPPPP',
'LPPPP',
'LPPPP',
'LPPPP',
'LPPPP',
'LPPPP',
'LPPPP',
'LPPPP',
'LPPPP',
'LPPPP',
'LPPPP',
'IPPPP']
aln_results.orthokmer_matrix.loc[[75, 113, 127, 157]].T
| 75 | 113 | 127 | 157 | |
|---|---|---|---|---|
| query_kmer | LPPPP | LPPPP | LPPPP | LPPPP |
| 9793_0:005123 | LPPPP | LPPPP | LPPPP | LPPPP |
| 1706337_0:000fc7 | LPPPP | LPPPP | LPPPP | LPPPP |
| 51337_0:001b5a | LPPPP | LPPPP | LPPPP | LPPPP |
| 9568_0:004ae1 | PPMPP | PPMPP | PPMPP | PPMPP |
| 43346_0:004190 | LPPPP | LPPPP | LPPPP | LPPPP |
| 885580_0:00488c | LPDRP | LPDRP | LPDRP | LPDRP |
| 10181_0:00305d | APSPP | APSPP | APSPP | APSPP |
| 1415580_0:000900 | LPPPP | LPPPP | LPPPP | LPPPP |
| 61221_0:00105a | LPPPP | LPPPP | LPPPP | LPPPP |
| 7897_0:0033c5 | LPPPP | LPPPP | LPPPP | LPPPP |
| 8407_0:002bff | LPPPP | LPPPP | LPPPP | LPPPP |
| 173247_0:004550 | LPPPP | LPPPP | LPPPP | LPPPP |
| 30732_0:0046dd | LPPPP | LPPPP | LPPPP | LPPPP |
| 241271_0:0048e4 | LPPPP | LPPPP | LPPPP | LPPPP |
| 8103_0:0045e4 | LPPPP | LPPPP | LPPPP | LPPPP |
| 56723_0:00152f | LPPPP | LPPPP | LPPPP | LPPPP |
| 210632_0:004c0c | LPPPP | LPPPP | LPPPP | LPPPP |
| 31033_0:00264e | LPPPP | LPPPP | LPPPP | LPPPP |
| 63155_0:004c86 | LPPPP | LPPPP | LPPPP | LPPPP |
| 7994_0:004d71 | LPPPP | LPPPP | LPPPP | LPPPP |
| 109280_0:00369f | LPPPP | LPPPP | LPPPP | LPPPP |
| 150288_0:004e5a | IPPPP | IPPPP | IPPPP | IPPPP |
Note
the k-mers are defined by position rather than sequence. You could easily make a variant of this method that uses the unique sequences instead. It would make the method slightly faster. The reason that I didn’t do this is because I wanted to mimic the LLM embedding version of Pairk, where identical k-mers have different embeddings and thus are treated as different k-mers.Inclusion of duplicate k-mers does alter the final z-scores, so it’s something to be aware of.
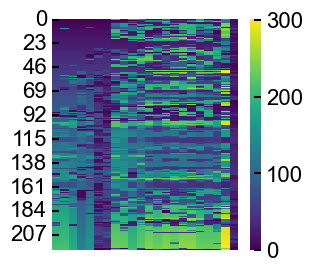
example: plot a heatmap of the matrices
import matplotlib.pyplot as plt
fig, ax = plt.subplots(figsize=(3,3))
aln_results.plot_position_heatmap(ax)
ax.xaxis.set_visible(False)
example: save the results to a file using write_to_file and load
them back into python using from_file:
aln_results.write_to_file('./aln_results.json')
aln_results = pairk.PairkAln.from_file('./aln_results.json')
print(aln_results)
PairkAln object for 222 query k-mers
query sequence: TNLGTVNAAAPAQPSTGPKTGTTQPNGQIPQATHSVSAVLQEAQRHAETSKDKKPALGNHHDPAVPRAPHAPKSSLPPPPPVRRSSDTSGSPATPLKAKGTGGGGLPAPPDDFLPPPPPPPPLDDPELPPPPPDFMEPPPDFVPPPPPSYAGIAGSELPPPPPPPPAPAPAPVPDSARPPPAVAKRPPVPPKRQENPGHPGGAGGGEQDFMSDLMKALQKKRGNVS
k-mer length: 5
Step 2: k-mer conservation
In this step, the query k-mer and the best matching homolog k-mers are
treated as a gapless multiple sequence alignment with ‘k’ columns, which
we call a “pseudo-MSA”. Column-wise conservation scores are calculated
for each position in each pseudo-MSA. All of the conservation scores are
then converted to z-scores to give the relative conservation of each
k-mer position compared to the rest of the query IDR. The conservation
score results are stored in a PairkConservation object which also
provides methods for plotting the results and reading/writing the
results from/to files.
calculate k-mer conservation - pairk.calculate_conservation()
the main method for Step 2 is the pairk.calculate_conservation()
method. It simply takes the pairk.PairkAln object as input, along with a
columnwise conservation scoring function and returns a
pairk.PairkConservation object.
The columnwise conservation scoring function can be any function that
takes a string of residues (a column of an alignment) as an input and
returns a float (conservation score). You can use custom functions here,
but pairk comes with a few built-in functions from Capra and Singh 2007
(DOI: 10.1093/bioinformatics/btm270) available in the
pairk.capra_singh_functions module. The
pairk.capra_singh_functions.property_entropy() is the default function
used by pairk.calculate_conservation().
Example
aln_results = pairk.pairk_alignment(
idr_dict=ex1.idr_dict,
query_id=ex1.query_id,
k=5,
matrix_name="EDSSMat50"
)
conservation_results = pairk.calculate_conservation(
aln_results,
)
example usage: using a different conservation scoring function:
from pairk import capra_singh_functions
column = 'NNNNNNNNNKNSNNNNNNNNSSN'
print(capra_singh_functions.shannon_entropy(column))
0.8161170118989496
aln_results = pairk.pairk_alignment(
idr_dict=ex1.idr_dict,
query_id=ex1.query_id,
k=5,
)
conservation_results = pairk.calculate_conservation(
pairk_aln_results=aln_results,
score_func=capra_singh_functions.shannon_entropy
)
k-mer conservation results - pairk.PairkConservation
The pairk.calculate_conservation() method returns a
pairk.PairkConservation object.
The returned pairk.PairkConservation object has matrices with similar
structure as pairk.PairkAln object matrices, except that they are numpy
arrays instead of pandas dataframes.
orthokmer_arr- the best matching k-mers from each homolog for each query k-mer - analogous to the orthokmer_matrix in thepairk.PairkAlnobjectscore_arr- the conservation scores for each position in the pseudo-MSA of each query k-merz_score_arr- the conservation score z-scores for each position in the pseudo-MSA of each query k-mer
If n is the number of k-mers in the query sequence and m is the
number of homologs (including the query sequence), the matrices will
have the dimensions:
orthokmer_arr: (n, m)score_arr: (n, k)z_score_arr: (n, k)
accessing the results
The row index of the arrays correspond to the starting position of the query k-mer in the query IDR.
For example, to access the conservation scores for the k-mer at position
4 in the query IDR, you would access the 4th row of the arrays:
.score_arr[4, :].
k_mer_position = 4
print(f"query k-mer at position {k_mer_position}: {conservation_results.orthokmer_arr[k_mer_position, 0]}")
print(f"pseudo-MSA for the query k-mer at position {k_mer_position} (including the query k-mer): {conservation_results.orthokmer_arr[k_mer_position, :]}")
print(f"scores for each position of the k-mer at position {k_mer_position}:")
print(conservation_results.score_arr[k_mer_position, :])
print(f"z scores for each position of the k-mer at position {k_mer_position}:")
print(conservation_results.z_score_arr[k_mer_position, :])
query k-mer at position 4: TVNAA
pseudo-MSA for the query k-mer at position 4 (including the query k-mer): ['TVNAA' 'TGNAA' 'TVNAA' 'TVNTA' 'TVNAA' 'TVNAV' 'TVNTA' 'TVSTA' 'NVNAN'
'AVSAG' 'TVSAS' 'SQNVA' 'TPNQA' 'TVKAK' 'PVNSF' 'PLNAL' 'TAAAA' 'TIKAK'
'TIKAS' 'TVKAK' 'TSNTS' 'TTAAA' 'NLNSQ']
scores for each position of the k-mer at position 4:
[0.69754401 0.48589838 0.6438038 0.64920359 0.44093655]
z scores for each position of the k-mer at position 4:
[ 0.17610372 -1.01357184 -0.12597394 -0.09562132 -1.26630561]
plotting the results
There are several plotting functions available from the
pairk.PairkConservation object shown below.
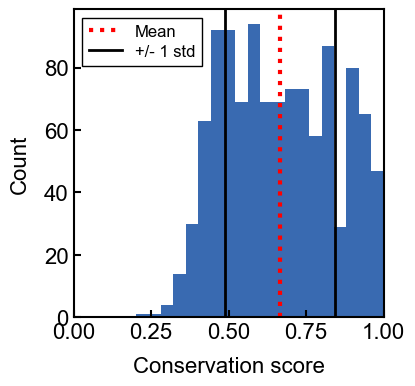
example usage: plotting background score distributions
ax = conservation_results.plot_background_distribution()
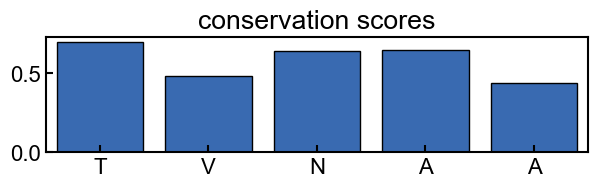
example usage: plotting conservation scores
fig, ax = plt.subplots(figsize=(7,1.5))
conservation_results.plot_score_barplot(k_mer_position, score_type='score', ax=ax)
ax.set_title('conservation scores')
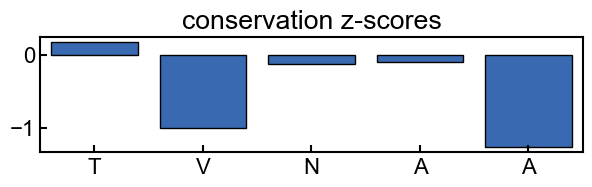
fig, ax = plt.subplots(figsize=(7,1.5))
conservation_results.plot_score_barplot(k_mer_position, score_type='z_score', ax=ax)
_ = ax.set_title('conservation z-scores')
example usage: display a pseudo-MSA as a sequence logo
fig, ax = plt.subplots(figsize=(7,1.5))
_ = conservation_results.plot_sequence_logo(k_mer_position, ax=ax)

fig, axd = conservation_results.plot_conservation_mosaic(
position=0,
score_type='z_score',
figsize=(8, 3)
)
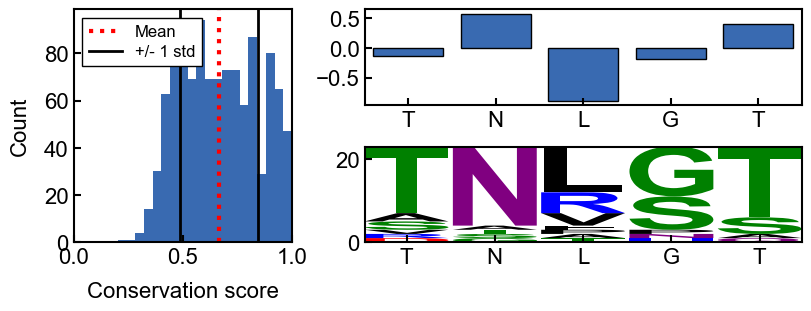
here is an annotated version of the mosaic plot:
annotated image
getting the average conservation score for a query k-mer
You can use the pairk.PairkConservation.get_average_score() function to get the average
conservation score for a k-mer position.
example: get the average conservation score for the query k-mer at position 4
conservation_results.get_average_score(4, score_type='z_score')
-0.4650737963634926
The pairk.PairkConservation.get_average_score() function takes a position_mask as an
optional argument that will only consider the conservation scores for
the positions in the mask when calculating the average score. This is
useful if you want to exclude certain positions from the average score
calculation.
example: get the average conservation score for the query k-mer at position 0, but only consider the conservation scores for positions 1, and 3 within the k-mer
position_mask = [0, 1, 0, 1, 0]
conservation_results.get_average_score(0, score_type='z_score', position_mask=position_mask)
0.197148283025409
You could also do a weighted average from manually extracted conservation scores.
example: get the weighted average conservation score for the query k-mer at position 0 (using some arbitrary weights)
import numpy as np
np.average(conservation_results.z_score_arr[0, :], weights=[0.1, 1, 0.5, 1, 10])
0.31155698039226354
Writing and Reading Results from Files
You can save the results to a file with pairk.PairkConservation.write_results_to_file() and load them back in with pairk.PairkConservation.read_results_from_file().
example usage: save the results to a file and load them back in
_=conservation_results.write_results_to_file('./conservation_results.npz')
conservation_results= pairk.PairkConservation.read_results_from_file('./conservation_results.npz')
advanced customization
Step 1 and Step 2 can be modified, but this requires a bit of knowledge
of the inner workings of the pairk package and the source code would
have to be modified directly and probably installed in editable mode
(see Getting Started). If you want to modify the package, here’s
roughly how I organized the code in the pairk directory (the source
code is available on github.
Below file paths are relative to the github repo root directory:
pairk/backend/- the main code for the package is located here. The main pairwise k-mer alignment and k-mer conservation functions are defined in files within this directory.pairk/__init__.py- User-facing functions are imported into the mainpairk/__init__.pyfile so that they are accessible when the package is imported. I think it also simplifies the import statements for users. Use this init file to find where pairk’s main functions are defined within the directory structure if you want to modify any of the functions above. You could also modify the init file to make any new functions you create easy to access.pairk/data/- data installed along with the package is stored here. This includes the scoring matrices and example data. The scoring matrices are stored in thepairk/data/matrices/folder.
The easiest customization to make would be to add a new scoring matrix.
To do this, you would add a new matrix file to the
pairk/data/matrices/ folder. The tools should be able to
automatically find the matrix file and add it to the available scoring
matrices. It will be named after the name of the file. Use
pairk.print_available_matrices() to confirm (make sure you’ve
installed pairk as an editable install for changes to take affect). You
could then use the new matrix in relevant methods by passing the matrix
name as an argument. If this doesn’t work, you may need to modify the
code that reads the matrices in the pairk/backend/tools/matrices.py
file.
!tree ../pairk/ -I __pycache__
../pairk/
├── __init__.py
├── _version.py
├── backend
│ ├── __init__.py
│ ├── conservation
│ │ ├── __init__.py
│ │ ├── capra_singh_functions
│ │ │ ├── __init__.py
│ │ │ └── capra_singh_2007_scores.py
│ │ └── kmer_conservation.py
│ ├── exceptions.py
│ ├── kmer_alignment
│ │ ├── __init__.py
│ │ ├── esm_embedding_distance.py
│ │ ├── needleman_tools.py
│ │ ├── scoring_matrix.py
│ │ └── scoring_matrix_needleman.py
│ └── tools
│ ├── __init__.py
│ ├── esm_tools.py
│ ├── matrices.py
│ ├── pairwise_tools.py
│ ├── plotting_tools.py
│ ├── pssms.py
│ └── sequence_utils.py
├── data
│ ├── README.md
│ ├── __init__.py
│ ├── example_alignment_9606_0_00294e-idraln-555_to_971-idr-440_to_665.fasta
│ ├── matrices
│ │ ├── BLOSUM62
│ │ ├── EDSSMat50
│ │ ├── __init__.py
│ │ ├── grantham.csv
│ │ ├── grantham_similarity_norm.csv
│ │ └── grantham_similarity_normx100_aligner_compatible
│ └── pairk_plotstyle.mplstyle
├── examples.py
├── py.typed
├── single_kmer
│ └── __init__.py
├── tests
│ ├── __init__.py
│ └── test_pairk.py
└── utilities.py
10 directories, 36 files